 The treatment and management options of COVID-19 patient are rapidly evolving. The amount of research published daily is endless so that keeping an overview seems almost impossible. This short review of current publications is intended to overview current treatment options and its evidence. We will look at: - How do you Identify and Triage Patients at Risk for Severe Disease? - What about High Flow Nasal Cannulas (HFNC) and Non-Invasive Ventilation (NIV)? - Should we Prone Position the Spontaneously Breathing Patient? - When to Use Corticosteroids? - Should we Use Remdesivir? - What about Convalescent Plasma? - How do we Manage Thromboprophylaxis? How do you Identify and Triage Patients at Risk for Severe Disease? In an ideal world, we would be able to assess newly admitted patients with COVID-19 to predict the risk of getting critically ill in the course of the disease. Apart from a proper clinical assessment, JAMA published the COVID-GRAM Risk Score to address this problem. They used a cohort of 1590 patients to develop this score and validated this with a cohort of 710 patients. From 72 potential predictors, ten variables were independent predictive factors and were included in the risk score. 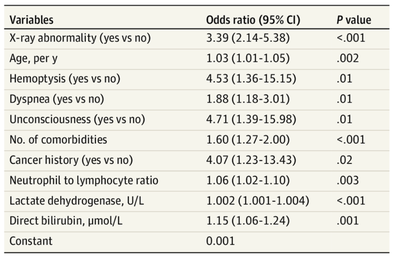 The practicability in a clinical setting is not clear yet, and as any predictive score, there are several limitations when it comes to assessing a single patient instead of a cohort. The COVID-GRAM Score Calculator can be accessed via the following link: http://118.126.104.170/ 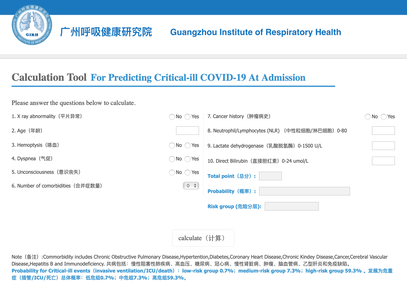 Early identification of COVID-19 patients at risk for severe disease would be helpful for management. Every clinic/ ICU should have a triage and risk assessment tool at hand.
For triage, we use the following simple criteria: 
As a predictive assessment tool for severe disease the COVID-GRAM Calculator can be used:
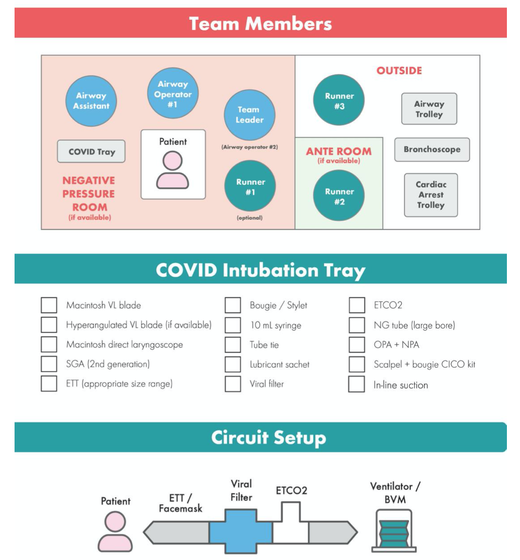
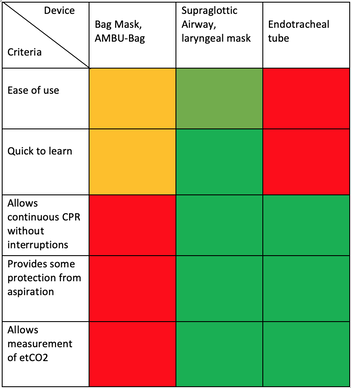
What about High Flow Nasal Cannulas (HFNC) and Non-Invasive Ventilation (NIV)? Especially at the beginning during the first wave of the pandemic, the use of HFNC and NIV was often avoided due to aerosolisation fear. Many ICU's tended to intubate their patients with respiratory failure relatively early. The lack of ventilators in some areas and reports that invasive ventilation is associated with high mortality (Zhou F, Lancet 2020; 395:1054) led to a constant change in management. KEEP IN MIND: Randomised-controlled studies for the treatment of COVID-19 patients with HFNC and NIV lack until now! Aerosolisation remains a big concern for health care workers (Niedermann MS; Am J Respir Crit Care Med 2020; 201:1019, Wu Z; JAMA 2020, February 24) and the amount of leakage flows is highly variable (Winck JC; Pulmonology 2020, April 20). Experience during the year 2020 showed, that most critical care providers have moved to use NIV and HFNC more frequently than initially. Proper personal protection equipment is essential and minimises risk for health care providers. Some evidence supports this approach (Avdeev SN, Am J Em Med AJEM Volume 39, p 154-157). NIV and HFNC is feasible in patients with COVID-19 and acute hypoxemic respiratory failure, even outside the ICU Helmet-NIV, leakage-free masks (non-vented masks) and double hose systems with virus-proof filters seem to be advantageous in this respect (Pfeiffer M; Pneumologie 2020, April 22). It is recommended that patients under HFNC should wear a surgical face mask over their cannulas Helmet NIV might advantageous compared to Mask NIV, though evidence is limited. (Patel BK et al. JAMA 2016. PMID: 27179847, single center study, trial stopped early, larger randomized-controlled studies awaited). KEEP IN MIND: Generally, there is only minimal evidence regarding the therapeutic benefit of these measures compared to their risks for the environment due to aerosolisation. Whether HFNC and NIV itself might produce self-inflicted lung injury (SILI) to some extend is not fully understood! Following patients should be considered for intubation and invasive ventilation - Severe hypoxemia (PaO2/FiO2 <150mmHg or respiratory rate >30/min) - Persistent or worsening respiratory failure (i.e. O2 sat <88%, RR > 36/min) - Neurologic deterioration - Intolerance of face mask or helmet - Airway bleeding - Copious respiratory secretions Should we Prone Position the Spontaneously Breathing Patient?Since the publication of Guerin C et al. (N Engl J Med 2013; 368:2159) prone positioning of patients with moderate to severe ARDS has become standard procedure in ICU's around the world. It is, therefore, evident that this treatment modality seems appropriate for COVID-19-induced lung injury, too. Trying to avoid intubations, clinicians rose the question, whether a prone position in the spontaneous breathing patient could avoid the need for invasive ventilation or even improve outcome. Ding L et al. (Crit Care 2020; 24:289) published a small multicenter study including 20 patients, whereas in 11 patients intubation could be avoided by prone positioning patients under NIV or HFNC. Telias et al. published an JAMA editorial (JAMA. 2020;323(22):2265-2267). He states that the prone position can improve oxygenation and can potentially result in less injurious ventilation. Unfortunately, this does not necessarily equate to lung protection and a better outcome. While improved oxygenation might prevent clinicians from intubating a patient, delayed intubation might worsen the patient's outcome. Regarding some evidence showing improved oxygenation during prone position, there are reasons to give it a try (Caputo ND et al. Acad Emerg Med Published online April 22, 2020). In the hypoxemic patient with no relevant respiratory distress awake prone positioning is a valid option - Use nasal cannulas or HFNC first - If comfortable enough, ask the patient to self-prone - Encourage the patient to remain in the prone position as long as well tolerated - Patients need close nursing and appropriate monitoring - Select prone positioning mattresses might be of help When to Use CorticosteroidsPatients with COVID-19 often show a biphasic course of the disease. The first phase is characterised by profound virus replication which decreases significantly after 5-7 days. After 7-10 days, a second phase develops in which an excessive or dysfunctional immune response can appear. This can lead to ARDS and multi-organ failure, which might be tackled by immunomodulating therapy. The largest, pragmatic randomised control trial we have at this stage is RECOVERY, performed in 176 hospitals around the UK and including more than 6400 patients (RECOVERY Collaborative group, N Engl J Med, July 17, 2020). COVID-19 patients that required oxygen or mechanical ventilation and presented with symptoms for at least seven days showed a significant reduction in 28-day mortality when treated with 6 mg Dexamethason OD for up to 10 days. Patients in the early viremic phase or patients that not required any oxygen performed worse with Dexamethasone. A broader insight into this topic brings a meta-analysis from JAMA in September 2020, including seven studies: DEXA-COVID19, CoDEX, RECOVERY, CAPE COVID, COVID STEROID, REMAP-CAP and Steroids-SARI. They ended up looking at 1703 patients and found a significant reduction in 28-day mortality when treated with steroids compared to placebo. Patients with COVID 19 that require oxygen, HFNC, NIV, mechanical ventilation or ECMO should be treated with steroids In patients not requiring oxygen, there is a trend towards harm when giving steroids - In these situations, steroids are NOT indicated Should we Use Remdesivir?Brief: Evidence in regards to the treatment with remdesivir is scattered and inconclusive. In the largest randomised control triad available so far is ACTT-1 looking at about 1600 patients (Beigel JH et al. N Engl J Med 2020; 383:1813-1826). In a few words, remdesivir showed a trend towards a 4-5 day shorter time to recovery, but not if symptoms existed for more than nine days. There was no significant influence on mortality, except maybe for patients requiring oxygen but not any help in ventilation. If at all, remdesivir might provide some advantage in a very selected patient group, but even this remains debatable. For this reason, many consider remdesivir the 'Tamiflu for COVID-19'. Two other papers remain to be mentioned briefly: Wang et al. (The Lancet; April 29) presented results from a relatively small study which was terminated early and showed no statistically significant clinical benefits of remdesivir - except for a trend towards a shorter duration of illness. Goldmann JD et al. presented the so-called '5 versus 10 days study', a phase 3 multicentre study with 397 patients. The primary outcome was their clinical status on day 14, secondary outcome patients with adverse events. Interestingly a 5-day course of remdesivir resulted in a better clinical outcome that a 10-day course. Again, It did not show any benefit compared to placebo. Remdesivir - The "Tamiflu for COVID-19" There is insufficient evidence to recommend the use of Remdesivir strongly. It is expensive, and if used, maybe there is only a short time window reasonable to act. Should We Use ECMO?During the early phase of the pandemic, first reports raised some concern that ECMO in COVID-19 patients might be associated with very high mortality (Henry BM et al. J Crit Care; 58:27). In the meanwhile, though we have new results from a more extensive cohort study looking at data from the Extracorporeal Life Support Organisation (ELSO, Barbaro RP et al. Lancet Volume 396, ISSUE 10257) The investigators looked at 1035 COVID-19 patients from 36 countries that were treated with ECMO (mean age 49 years, 74% male). 70% of all patients had relevant co-morbidities. The median time of ECMO support was 14 days. The incidence of in-hospital mortality 90 days after the initiation of ECMO was 37·4%. Mortality was 39% in patients with a final disposition of death or hospital discharge. These results are comparable with earlier mult-centre studies with patients suffering from non-COVID-19 ARDS (Combes A et al. N Engl J Med 2018; 378:1965). A retrospective cohort study from France looking at 83 patients treated with ECMO showed a probability to die after 60 days of 31%. Mortality at the time of the last follow-up was 36% (Schmidt et al. Lancet Respir Med 2020; 8:1121-1131). Various Societies recommend the use of ECMO in COVID-19 patients with treatment-refractory lung failure (Surviving Sepsis Campaign, ESICM, SCCCM and ELSO, WHO) Regarding the ongoing pandemic and limited resources, uniform indication and selection criteria for ECMO use should be available What about Convalescent Plasma?After a negative small randomised control trial (Li L et al. JAMA. 2020;324(5):460-470), a controversial Emergency Use Authorisation was granted by the FDA on 23.8.20 due to an observational study with a favourable effect on mortality with a high specific IgG content and onset less than days after symptom onset (Joyner MJ et al. MedRxiv; https://doi.org/10.1101/2020.08.12.20169359 - non peer-reviewed). At this stage the use of covalescent plasma can not be recommended How do we Manage Thromboprophylaxis?COVID 19 undoubtedly causes an inflammatory state that seems to trigger thrombotic activation in the venous and the arterial circulation. Thromboembolic complications are common, but the evidence is not robust on whether prophylactic or therapeutic doses should be used. Patients often have a significant elevation of D-dimers, an acute phase reactant representing the severity of disease rather than the dosage of thromboprophylaxis. One observational study looking at 1716 patients found no improved outcomes among in-hospital patients with COVID-19 when treated with therapeutic anticoagulation compared to prophylactic dosing. Moreover, patients who were started on anticoagulation for COVID-19 without evidence of thrombosis, new VTE, or new atrial fibrillation had worse outcomes compared to patients who were on prophylactic anticoagulation (Patel NG et al. Thrombosis Update; Volume 2, 2021, 100027) A case-based review of current literature and the COVID-19 specific coagulopathy end with the same finding that all in-hospital patients should receive prophylactic thromboprophylaxis. Whether a higher dose of prophylactic anticoagulation may be more effective is currently unknown (Chen EC et al. Oncologist. 2020 Oct; 25(10): e1500–e1508.). A small and retrospective study with 152 patients showed a lower risk of death and a lower cumulative incidence of thromboembolic events in patients with respiratory failure when a high-dose thromboprophylaxis was used. (Jonmarker S et al. Critical Care volume 24, Article number: 653 (2020)). Evidence supports the use of prophylactic thromboprophylaxis in patients with COVID-19 Whether a higher dose of anticoagulation might be more effective is currently unknown This single slide turn you into an expert in the nomenclature of monoclonal antibodies, but also helps to understand quickly what sort of medication your patient is treated with. Share and Care!   The Aerosol-Danger of SARS-Cov-2The outbreak of the SARS Coronavirus-2 (SARS-CoV-2) in China 2019 has within a short time spread around the globe and is just about to hit central Europe. Although about 80% of all confirmed cases develop a mild febrile illness, around 17% develop severe Corona viral disease (COVID-19) with findings of acute respiratory distress syndrome (ARDS), of which about 4% will require mechanical ventilation. Since this virus, which was previously unknown to humans, spread rapidly around the globe, a large number of patients requiring intensive medical care now arise within a very short time. The lungs are the organs most affected by COVID-19 because the virus accesses host cells via the enzyme ACE2, which is most abundant in type II alveolar cells of the lungs. This results in mainly type 1 respiratory failure, which often requires urgent tracheal intubation and mechanical ventilation. Due to viral shedding in the patient's lungs, COVID-19 spread mainly via droplets. Events like coughing, high flow nasal oxygen (High-Flow), intubation and more can cause aerosol generation, allowing these airborne particles to travel even further distances. Performing endotracheal intubation in these patients is, therefore, a high-risk procedure, and it is required to adhere to certain principles to avoid infection of health care providers. The Safe Airway Societies of Australia and New Zealand have published a consensus statement that describes the problem very well and provides practical tips based on the currently available evidence. 1. Non Invasive Ventilation (NIV) and High Flow Nasal Oxygen (High-Flow)Current evidence suggests that the failure rate of NIV in COVID-19 patients seems to be similarly high as observed among Influenza A patients. Failure in these patients resulted in higher mortality. In general, NIV is recommended to be avoided or at least used very cautiously! The utility of High-Flow in viral pandemics in unknown. There is some evidence suggesting a decreased need for tracheal intubation compared to conventional oxygen therapy. High Flow Nasal Oxygen is worth a try, although it has to be assumed, that this is aerosol-generating. High-Flow should only be used in (negative pressure) airborne isolation rooms, and staff should wear full personal protective equipment (PPE) including N95/P2 masks. NIV and High-Flow are NOT recommended for patients with severe respiratory failure or when it seems clear that invasive ventilation is inevitable!  Full Personal Protective Equipment PPE
|
||||||






| Pocket Cards Toxine und Antidots |



Acute decompensated heart failure comes in many ways as remains a challenge for optimal treatment since various conditions can cause it, e.g. advanced chronic heart failure, cardiogenic- and septic-shock, cardiac- and non-cardiac surgery, etc. Among many other interventions, the infusion of positive inotropes is often used on one side and vasodilators on the other side to stabilize the situation and improve cardiac function. Despite all these measures, the re-admission rate and mortality remain a significant problem among these patients.
While Levosimendan is still under investigation in the U.S., it is used worldwide for the short-term treatment of acutely decompensated severe chronic heart failure, especially when other treatment options have failed. Some recommend the usage of this drug in a different setting like sepsis-related heart failure or coronary heart disease.
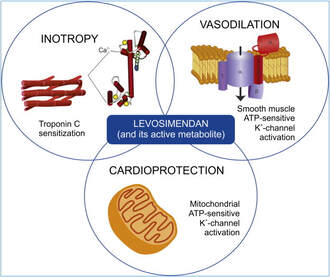
How does Levosimendan work?
Levosimendan has some remarkable properties, which are mainly caused by three mechanisms:
1. Enhancement of the calcium sensitivity of the myofilament by binding to troponin C.
2. Opening of adenosine triphosphate-sensitive potassium (KATP) channels in vasculature smooth muscle.
3. Opening of mitochondrial KATP channels.
These mechanisms result in a positive inotropy of the heart, an increase in stroke volume (SV) and therefore, cardiac output (CO). Besides, it's vasodilatory properties seems beneficial for coronary perfusion and reduce pulmonary capillary wedge pressure (PCWP).
All these effects do not appear to induce an unfavourable energy balance in the myocardial cell, and also the oxygen demand is not increased.
So what's the Evidence?
The Small Bits and Pieces
First publications back in the 90ies and early 2000s were indicative for the properties of levosimendan. Three studies were randomized and double-blinded but very low in patient numbers and therefore not powered enough to provide substantial evidence. Their statements were also not concerning patient outcome or mortality, but its results confirmed that levosimendan enhances cardiac output without oxygen wasting, is well tolerated and leads to favorable hemodynamic effects.
Lilleberg et al. Eur Heart J. 1998;19:660–668.
Ukkonen et al. Clin Pharmacol Ther. 2000;68:522–531.
Nieminen et al. J Am Coll Cardiol. 2000;36:1903–1912.
In 2003 Kivikko et al. published further data from another small trial indication that its beneficial effects (decreases in left and right heart filling pressures and in SVR, as well as increases in stroke volume and cardiac index) are maintained for at least 24 hours after discontinuation of a 24-hour infusion. At this point, it is worth mentioning that the author published these findings as a current employee of Orion Pharma, which manufactures levosimendan.
Kivikko et al. Circulation. 2003;107:81–86.
Further data indicated that its effect might be sustained for up to at least a week, although also this study included only 22 patients!
8. Lilleberg et al. Eur J Heart Fail. 2007;9:75–82.
A Lancet publication in 2002 by Follath et al. presented a multicentre, randomized, double-blind trial in which they compared the effect of levosimendan to dobutamine in patients that were admitted to a hospital with low-output heart failure and were judged to require hemodynamic monitoring and treatment with an intravenous inotropic agent. In these 203 patients, levosimendan had a consistently better effect than dobutamine on the individual hemodynamic variables at the end of the 24 h treatment period (cardiac output and pulmonary capillary wedge pressure). The change in clinical symptoms like fatigue and dyspnoea were not significantly different, though.
Interestingly, for the first time, the levosimendan group showed lower mortality than in the dobutamine group for up to 180 days. However, it's important to mention its absence of placebo control and its rather small sample size.
Follath et al. Lancet. 2002;360:196–202.
The Big Lumps
The SURVIVE Study
In the SURVIVE study Mebazaa et al. compared the efficacy and safety of intravenous levosimendan or dobutamine in patients hospitalized with acute decompensated heart failure who required inotropic support.
randomized, double-blind trial at 75 centers in 9 countries
in which they evaluated
1327 patients hospitalized with acute decompensated heart failure who required inotropic support
They found that
the addition of levosimendan to standard therapy resulted in fewer deaths compared with dobutamine, especially in the first few weeks after treatment
but
Levosimendan did not significantly reduce all-cause mortality at 180 days or affect any secondary clinical outcomes
The REVIVE Studies - Heart Failure
In the Randomized Multicenter Evaluation of Intravenous Levosimendan Efficacy studies (REVIVE I and REVIVE II) the efficacy of levosimendan on symptoms of heart failure during five days after starting a 24h trial drug infusion was assessed.
Each patient's clinical course over 5 days was determined by a composite of the patient's self-assessment of symptoms together with a physician's assessment of the occurrence of clinical deterioration.
The primary endpoint of the study was a new clinical composite endpoint (improvement, unchanged or worse - including death) derived from studies in chronic heart failure and first evaluated in 100 ADHF patients in the REVIVE I pilot study.
first large, prospective, randomized, double-blind, controlled trials
in which they evaluated
600 patients admitted at 103 sites in the United States, Australia, and Israel
with
worsening heart failure and dyspnea at rest despite treatment with intravenous diuretics, and left ventricular ejection fraction of < 35% measured within the last year
whereas
patients were randomized to either receive levosimendan for 24h or standard treatment alone
They found that after 5 days
- more patients receiving levosimendan experienced improvement compared with those who were on placebo (19.4% vs 14.6%, respectively, a 33% relative increase; P = .015)
- fewer patients receiving levosimendan worsened compared with patients who were on placebo (19.4% vs 27.2%, respectively), a 29% relative decrease, and
- fewer patients receiving levosimendan required rescue therapy (15.1%) vs placebo (26.2%), a 42% relative decrease.
Among other secondary endpoints, levosimendan improved
B-type natriuretic peptide (BNP) levels, length of hospital stay and dyspnoea at 6 hours
But
mortality at 90 days did not differ significantly between treatment arms (a secondary endpoint) and
the most common treatment-emergent cardiovascular adverse events were more frequent with levosimendan, including hypotension (50% vs 36%), ventricular tachycardia (25% vs 17%), and atrial fibrillation (8% vs 2%).
In this study investigators wanted to know whether in adult patients who have sepsis the application of levosimendan reduces the incidence and severity of acute organ dysfunction compared with placebo.
Randomised, double-blind, placebo-controlled multi-centre trial in 34 general ICUs in the UK
in which they evaluated
516 patients with suspected or confirmed infection and 2 or more SIRS criteria who were dependent on vasopressors for at least four hours to maintain their blood pressure.
They compared
the intravenous infusion of levosimendan or placebo for 24 hours in addition to standard care
and found:
- No significant difference in the daily Sequential Organ Failure Assessment (SOFA) score up to day 28 (primary outcome)
- No statistical difference on death at 28 days, at ICU discharge and hospital discharge (secondary outcome)
- And: The use of levosimendan was associated with more hemodynamic instability, lower mean arterial pressures, therefore more need for noradrenalin at 24h and significantly more supraventricular tachyarrhythmias (all secondary outcomes).
The CHEETAH Trial - Cardiac Surgery
The question in CHEETAH was if levosimendan compared to placebo reduces mortality in patients undergoing cardiac surgery with left ventricular dysfunktion.
This time Landoni et al. performed a
Multi-centre, randomised, placebo-controlledand parallel group designed trial
in which they evaluated
506 patients scheduled for cardiac surgery with peri-operative cardiovascular dysfunction
defined as
pre-operative left ventricular ejection fraction (LVEF) < 25%, pre-operative intra-aortic balloon pump (IABP), intra- or post-operative (within 24 hours) IABP or significant inotropic requirement
They compared
Levosimendan infusion continued for up to 48 hours (or until ICU discharge) or placebo
and found
No difference in mortality at thirty 30 days (primary outcome)
And no difference in survival over time, renal replacement therapy, the median duration of mechanical ventilation, the median hospital stay and interruptions due to adverse events (all secondary outcomes).
In contrast to these clinically somewhat discouraging results Shang et al. have published a meta-analysis in 2017 of randomized controlled trials looking at the usage of levosimendan in patients with heart failure, cardiogenic shock and acute coronary syndrome.
They ended up looking at a total of nine studies, most of them low in patient numbers and comparing levosimendan to either placebo or other drugs (dobutamine and enoximone).
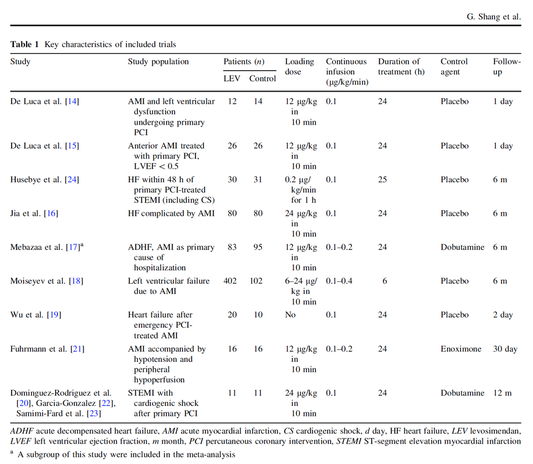
According to these data the authors conclude that levosimendan is associated with reduced total mortality, decreased incidence of worsening HF, and improved hemodynamic outcomes and does not increase the risk of adverse events except for hypotension in patients with HF (including CS) complicating ACS. Thus, levosimendan should be recommended for routine clinical application in these patients.
Am J Cardiovasc Drugs 2017 Dec;17(6):453-463.

According to current evidence
- Levosimendan has interesting mechanisms of action and successfully seems to enhance cardiac output without additional oxygen wasting. This includes decreased right and left ventricular filling pressures, decreased SVR, as well as increases in stroke volume and cardiac index. Also, PCWP is decreased.
- In terms of various patient groups, patients with acute heart failure or acute on chronic heart failure appear to benefit in terms of clinical symptoms, if at all.
- Unfortunately, there is no convincing clinical evidence that levosimendan has any benefit in long term outcomes in terms of mortality.
- And, levosimendan seems to be associated with some potential treatment-emergent cardiovascular adverse events like hypotension, supraventricular and ventricular arrhythmia.
Acute myocardial infarction - It's pain radiating to the right arm we have to worry about!
15/3/2019

A recent publication in the BMJ shows, that positive troponin levels are found frequently in patients with non-cardiac problems. This finding underlines the importance of good history taking, including the assessment of chest pain (CP) characteristics.
The TRAPID-AMI Study
They performed an
international, multicenter diagnostic study
in which they evaluated
1282 patients admitted for possible AMI to emergency departments in Europe, the US and Australia
The outcome was
the correlation of symptoms with the diagnosis of AMI and its relation to myocardial infarction size
They found that
1. only 4 symptoms were independently predictive of AMI
- Radiation to the right arm/ shoulder (OR 3.0, CI 1.8-5.0)
- Chest pressure (OR 2.5, CI 1.3-4.6)
- CP worsened by physical activity (OR 1.7, CI 1.2-2.5)
- Radiation to left arm/ shoulder (OR 1.7, CI 1.1-2.4)
2. Patients with more than 1 of the 4 symptoms were more likely to have AMI
- For patients who had all 4 symptoms, 55% had a diagnosis of AMI
3. Patients with larger AMI's were more likely to have
- pulling CP
- pain in the right upper chest (right supramammillary area)
- and right arm/shoulder radiation

- It's time to get rid of the classical image of CP radiating only into the left arm. While this might still be the predominant complaint at presentation, it's the right sided shoulder/ arm pain we should keep a close eye on!
- Chest pain radiating to the right shoulder/ arm is more predicitive of myocardial infarction than left sided chest pain
- And remember: a positive Troponin alone does not fullfill the diagnostic criteria of AMI!
Crit Pathw Cardiol. 2019 Mar;18(1):10-15.
Top Ten Websites in Critical Care Medicine Education

The Journal of Intensive Care Medicine has recently published a review of websites providing educational content that is part of the Free Open Access Meducation (FOAMed) movement. The authors have searched the web for critical care medicine education websites and have identified 97 sites they consider relevant for critical care.
These websites were then reviewed and evaluated using the Critical Care Medical Education Website Quality Evaluation Tool (CCMEWQET). They were then split up into three tertiles according to their score.
Congratulations to the Top Ten websites that indeed provide excellent educational information freely accessible.
Oh, and by the way we might mention that our small site Crit.Cloud has been considered among 96 other as relavant in this field and has ended up in the middle tertile with its ranking. 🙃
A big thank you to the authors of this paper for their excellent work and for providing us with an updated list of websites worth taking a closer look! We are delighted to follow their wish by sharing the table of these websites.
Wolbrink TA et al. J Intensive Care Med. 2018 Jan;34(1):3-16
Full List of all Website Reviewed (Click on a table enlarge)






On arrival to our ICU, the patient was noted to be still unresponsive. He showed no reaction to painful stimulation. The Pupils were rather wide, but symmetrical in size and showed a prompt response to light. Brain stem reflexes were present and peripheral reflexes were triggerable. His breathing pattern was regular, and saturation levels were above 96% on room air. A blood gas analysis showed a normal pH and normal CO2-levels.
As usual, the common reflexes started to kick in, and some suggested to go for a CT-scan of his head to out-rule some significant complications. Luckily enough, close observation revealed some slow improvement of his alertness, and we considered the possible diagnosis of a central anticholinergic syndrome (CAS).
After the application of 1mg of physostigmine (2x0.5mg) intravenously the patient almost promptly awoke and had an uneventful stay on our unit.
This case just reminded me of these many patients in anaesthesia that inexplicably show delayed awakening after sedation or a general anaesthetic.
In fact, it is estimated that the incidence of central anticholinergic syndrome is around 8- 12 % following general anaesthetic and lesser with regional anaesthesia.
What is a Central Anticholinergic Syndrome?

Classically the central anticholinergic syndrome (CAS) describes a condition where a substance causes a competitive antagonism of acetylcholine (ACh) at peripheral and central muscarinic receptors. Initially, these were plants containing atropine, hyoscyamine and scopolamine.
There are four muscarinic receptors:
- M1 mainly in the central nervous system (responsible for delirium when antagonised)
- M2 in the brain and heart
- M3 in the salivary glands and
- M4 in the brain and lungs
The PERIPHERAL SYNDROME presents with:
- Dry mouth
- Difficulty swallowing (lack of saliva)
- Photophobia and blurred vision (due to dilated pupils)
- Dry skin, fever
- Reduced bowel sounds and urinary retention
The CENTRAL SYNDROME presents with:
- Agitation, agitated delirium, visual and auditory hallucinations
- Hypoactive delirium may also occur, this seems to be more common though in the postoperative setting
The clinical diagnosis of a CAS is more straightforward when typical peripheral symptoms accompany central signs.
The Problem with the "Silent" Postoperative CAS
The clinical diagnosis of a CAS is often straight forward when typical central symptoms accompany peripheral symptoms. The problems are patients in the postoperative setting, in which the patient often presents with somewhat atypical central symptoms and often minimal or even no peripheral symptoms at all.
Especially in this setting, the anticholinergic syndrome may be accompanied by sedation or coma. The mechanisms causative for this phenomenon are not well understood but might include greater tolerance at peripheral receptors, longer persistence at central receptors or greater CNS susceptibility due to age or disease.
Postoperative CAS is often associated with atypical central symptoms and minimal or even no peripheral signs at all! Sedation and coma are often observed in this setting.
What Drugs Cause CAS?
Common anticholinergics agents should be more accurately referred to as antimuscarinics, as these agents do not generally block nicotinic receptors.
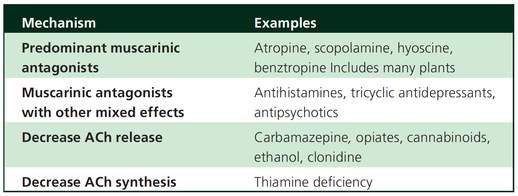
The Problem is that many currently used drugs in anaesthesia and critical care are also known to cause this syndrome.
This includes:
Benzodiazepines, opioids, phenothiazines, butyrophenones, ketamine, etomidate, propofol, nitrous oxide, and volatile inhaled anaesthetics!
How Can I Diagnose a "Silent" Postoperative CAS?
The fact that postoperative CAS often lacks the presence of peripheral symptoms makes the diagnosis challenging. The different presentation of the syndrome ranging from somnolence, confusion, amnesia, delayed recovery, stupor, coma to agitation, hallucinations, dysarthria, ataxia, delirium makes it difficult to diagnose accurately.
It is, therefore, a diagnosis of exclusion! The most helpful tool you have is physostigmine!
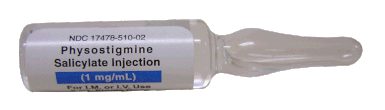
The prompt arousal of a patient after the application of intravenous physostigmine is highly suggestive of a postoperative central anticholinergic syndrome.
How to Use Physostigmine
When dealing with prolonged somnolence or unexplained agitation following any form of anaesthesia, make sure to check and monitor vital signs and provide basic or advanced life support if necessary.
Exclude common causes first (e.g. overhang of sedatives or opioids, persistent muscular paralysis, hypoxemia, hypercapnia, hypoglycemia and other).
If common causes can be excluded and CAS is a probable diagnosis, you should consider the application of intravenous physostigmine.
Physostigmine in recommended doses is considered safe! Possible adverse effects are considered unlikely. Studies found cholinergic symptoms sometimes to be mild, and these adverse effects are more an indication of probable excessive doses rather than an established safety concern.
For postoperative CAS a dose of 1mg of physostigmine i/v is recommended.
This dose can be divided into two doses of 0.5mg i/v if required.
The maximum dose recommended in this setting is 2mg i/v.

Beware: You are giving Physostigmine Salicylat - Do not give to patients with aspirin allergy!


So if you work in an emergency room, anaesthesia or intensive care, there's a good chance you will be facing an anticoagulant patient with potentially critical bleeding that could require urgent treatment... And this leaves you with the following questions:
- What is a critical bleed (apart from obvious massive bleeding)? Does this bleeding need imminent reversal?
- Do I need any laboratory testing before?
- What treatment should I actually give the patient?
If you do not have a guideline in your institution, it may be time to create one, and the following publication is indeed very useful for this purpose!
The 2017 ACC Expert Consensus Decision Pathway on Management of Bleeding in Patients on Oral Anticoagulants very nicely summarises current evidence and expert opinion on these issues. But the very best are their excellent figures, providing all the answers you need: simple and very understandable!
What is a Critical Bleeding?
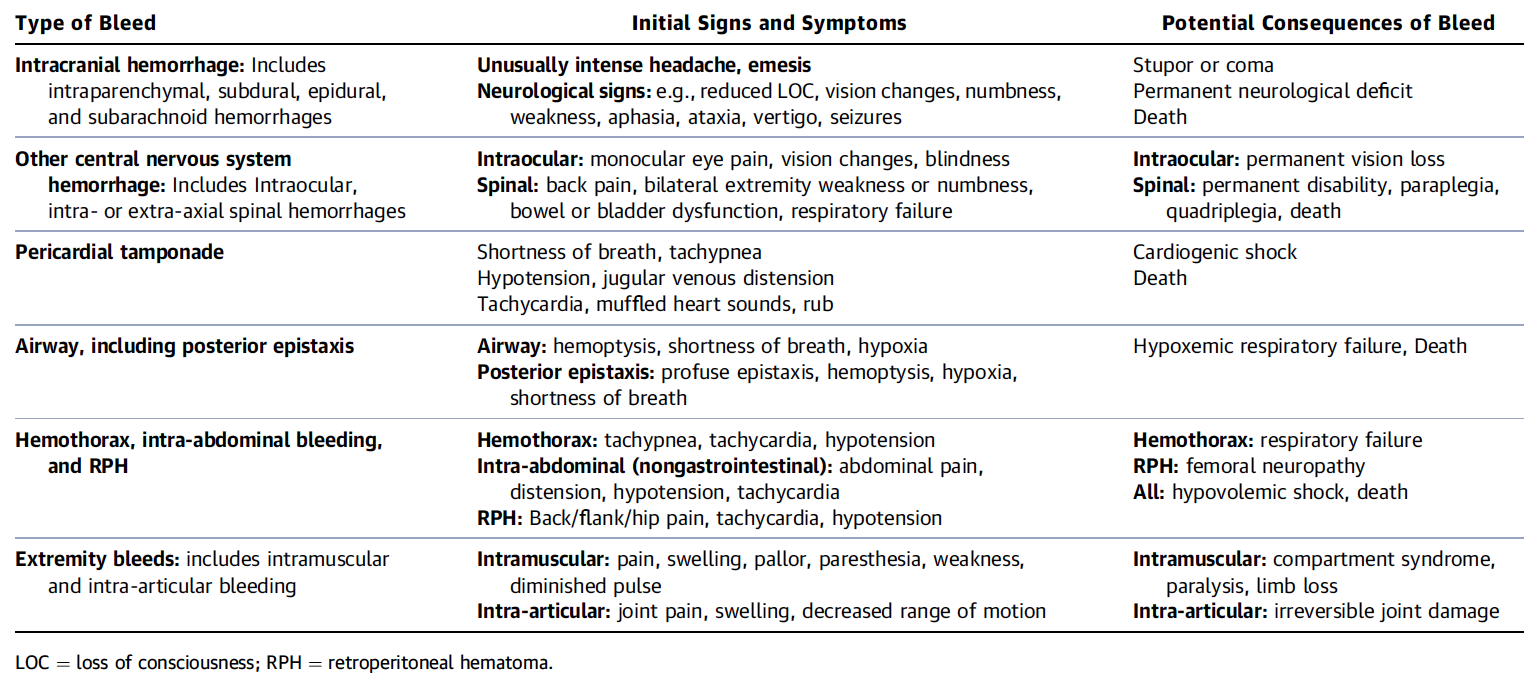
Do I Need any Laboratory Tests Before?

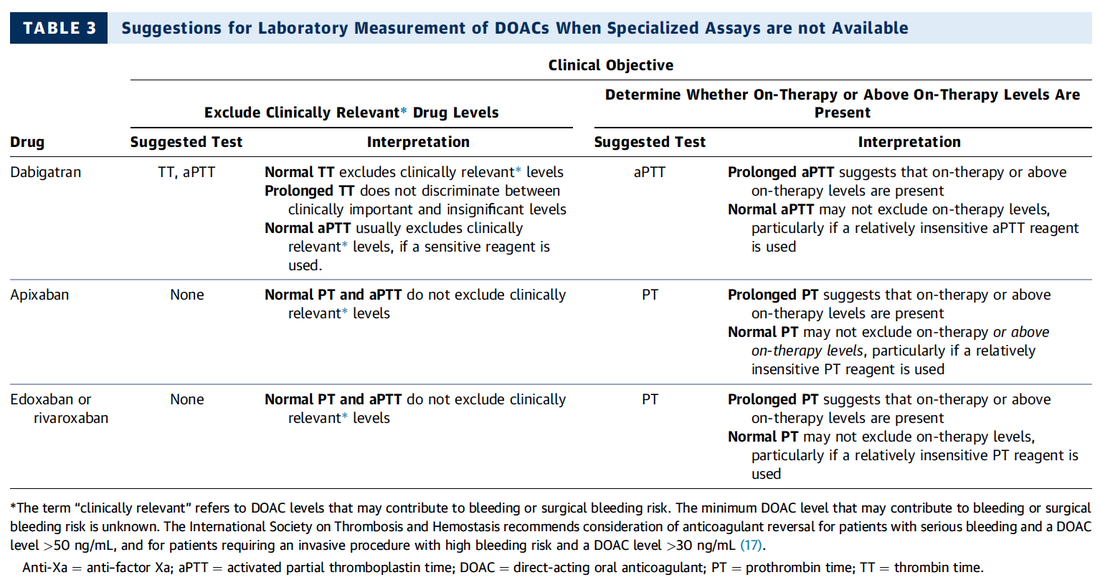
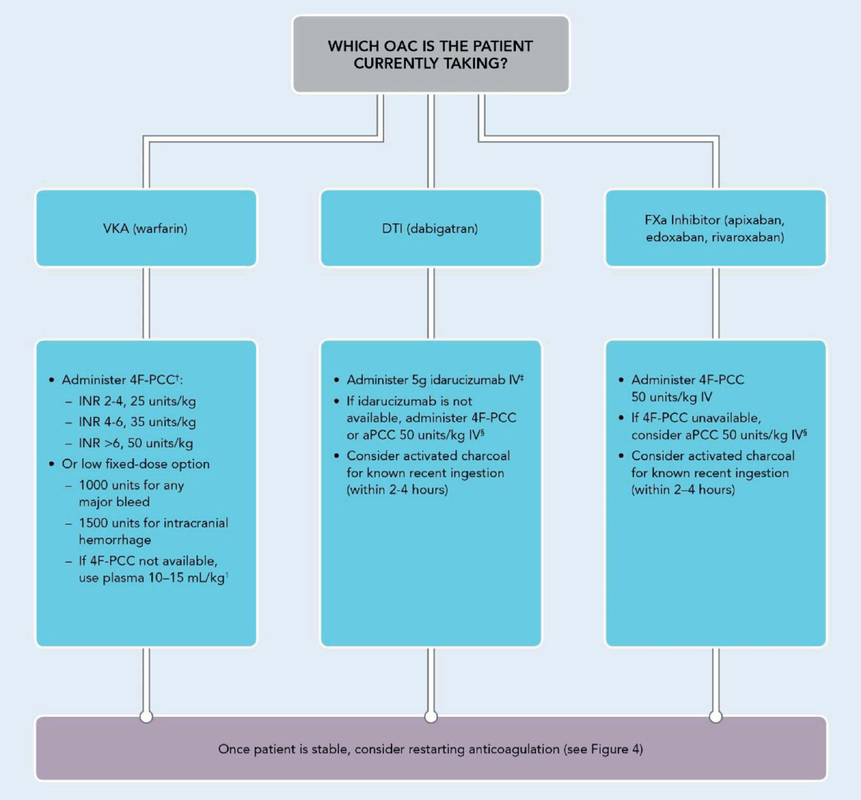
What Treatment Should I Give the Patient for Reversal?

Interestingly a discussion started on whether giving Lasix as a first line agent in the acute setting of pulmonary oedema is beneficial or not. A quick look into to current literature gave no clear answer and reading further into the topic revealed unusual properties of Lasix we hadn't been really aware of so far. We all use and love Lasix, but do we really know the drug?
The Beginning of Lasix
Furosemide (sometimes also called frusemide) was first developed by 'Farbwerke Hoechst AG' in Frankfurt am Main, Germany, a company that was founded back in the year 1863. Karl Stürm, Walter Siedel and Rüdi Weyer set the basis with the invention of N-substituted-3-Carboxy-6-Halo-Sulfanilamide, and it's derivates, one of them being furosemide. The researchers soon noticed its saluretic (sodium Na, potassium K and chloride Cl) and diuretic effect in almost equivalent proportions. As these substances did not cause any acidosis nor alkalosis, they suggested their future use for the treatment of oedema and hypertension.
Researchers soon noticed that the diuretic effect of furosemide lasted for about 6 hours... 'LAsts for SIX hours'... and therefore gave it the name: LASIX!
What is Furosemide
Furosemide is an organic anion from the group of loop diuretics (as are bumetanide and torasemide) and is sold under the brand name of Lasix©. Its indications are for the treatment of oedema due to heart or liver disease as well as kidney disease. It is also used for the treatment of mild or moderate hypertension. Furosemide has become one of the cornerstones in the treatment of heart failure.
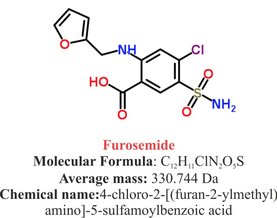
Furosemide can be applied by oral intake as a tablet or as an intravenous injection. Once in the bloodstream, it is predominantly bound to proteins (>90%).
Loop diuretics do not undergo glomerular filtration. In fact, they pass the glomerulus and are actively secreted across proximal tubular cells by organic anion transporters and the multidrug resistance-associated protein 4 (area A). It is important to know that non-steroidal anti-inflammatory drugs (NSAID) and endogenous uremic anions compete with this loop diuretic secretion and can cause 'diuretic resistance'.
Once loop diuretics have reached the tubular system, they bind to sodium-potassium-chloride co-transporters (NKCC2) in the ascending limb of the loop of Henle and block the reabsorption of these ions directly (area B). Further down at the macula densa they inhibit the same co-transporter (area B) thereby stimulating renin secretion and inhibiting tubuloglomerular feedback. This results in preserved glomerular filtration despite increased salt delivery to the macula densa. All this finally results in the loss of sodium, chloride and potassium and therefore loss of water.
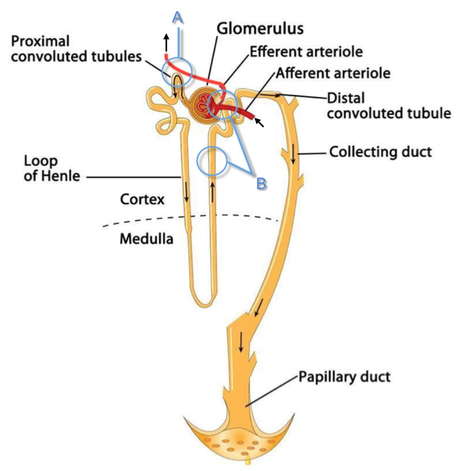
Furosemide also interacts with other sodium-potassium-chloride co-transporters (NKCC1) elsewhere in the body:
- Blocking NKCC1 in the ear probably explains the ototoxicity of loop diuretics
- Blocking NKCC1 in smooth muscle cells causes vasodilation
- Blocking NKCC1 in the afferent arteriole and near the macula densa elevates renin secretion and the generation of angiotensin II
These complex interactions on haemodynamics explain that the net response in each patient might be different. On the one hand, loop diuretics dilate blood vessels directly and increase the level of vasodilatory prostaglandins. On the other hand, some of these effects counteract each other making it difficult to predict which effect will finally predominate.
Many studies have looked closer into the vasoactive properties of furosemide. Current evidence indicates that it has a systemic venodilator effect which actually reduced preload acutely. The same investigators found a reduction in the right atrial pressure and the pulmonary capillary wedge pressure, presumably reflecting the systemic venodilator effect of furosemide.
While the acute venodilator effect may be beneficial to the failing heart, its action on arteries might be detrimental. Several studies have shown that in patients with chronic heart failure furosemide causes arterial vasoconstriction. Also, there is one study showing that pulmonary vascular resistance in healthy volunteers rose significantly.
Francis GS et al. described how the administration of furosemide actually led to decreased LV function, increased LV filling pressures, increases in MAP, SVR, plasma renin activity, and plasma noradrenaline levels.
Beneficial venodilator response predominates over arterial vasoconstriction in patients with (1) myocardial infarction and (2) salt depleted volunteers.
Venous relaxant effect has not been demonstrated in patients with chronic heart failure. In this setting detrimental arterial vasoconstriction seems to predominate.
Pardeep S et al. Br J Clin Pharmacol. 2000 Jul; 50(1): 9–13.
Francis GS et al. Ann Int Med 1985; 103(1): 1-6.
Pharmacological Properties
Administered furosemide orally has a limited and highly variable bioavailability. The diuretic effect starts within the first hour, and the duration of action is around 6 hours (4-8 hours). Injected furosemide intravenously is approximately twice as potent on a per-milligramme basis as oral doses.
In acute decompensated heart failure sodium retention becomes more avid and higher peak levels might be required to become more effective. This can be achieved by giving furosemide intravenously.
Once a loop diuretic is administered, the excretion of sodium chloride is increased for several hours. This is then followed by a period of very low sodium excretion resulting in a so-called 'post-diuretic retention'.
How to use Furosemide for Acute Decompensated Heart Failure (ADHF)
So far for the basics of furosemide, but what about its usage for acutely decompensated heart failure? Should furosemide be given as soon as possible or not?
The 2013 ACCF/AHA guidelines for the management of patients with heart failure give diuretics a class I recommendation. The evidence behind these recommendations though is level B or level C only! So these recommendations are not really helpful to answer this question.
The authors in UpToDate® mention diuretics directly after the use of oxygen. For patients with evidence of volume overload their recommendation is to give loop diuretics immediately (Grade 1B) as there is evidence that in this setting this may improve outcomes. They also suggest that patients with ADHF usually are volume overloaded, therefore indicating that most patients should receive diuretics ASAP.
The only exception they mention where some delay in inducing diuresis might be required is in patients with severe hypotension or cardiogenic shock.
There is reasonable doubt that patients with ADHF are usually volume overloaded, as suggested by UpToDate®. Zile MR et al. demonstrated that while most patients with acute pulmonary oedema have increased filling pressures, most did not have significant increases from their dry weight on presentation! Fallick et al. actually argue that it isn't fluid gain but rather shift in fluids from other compartments, mainly shift from the splanchnic circulation, which usually is very compliant.
And as mentioned above, there is evidence that giving a straight shot of furosemide might actually influence haemodynamics negatively in different ways (decreased LV function, increased LV filling pressures, increases in MAP, SVR, plasma renin activity and plasma noradrenaline levels).
In conclusion there is no straight forward answer to this question, but I would put it down as follows:

- Furosemide should not be routinely used for the immediate treatment of acute decompensated heart failure (ADHF)/ acute pulmonary oedema
- However, in patients with evidence of volume overload the administration of early furosemide (preferentially given as an intravenous bolus) seems beneficial and improves outcome. But beware, most patients are not volume overloaded!
- In urgent situations the focus should be on early non-invasive ventilation and the administration of nitroglycerin!
Wilson S et al., UpToDate.com 2018
WRITING COMMITTEE MEMBERS, Yancy CW, Jessup M, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128:e240.
Zile MR, Bennett TD, St John Sutton M, et al. Circulation 2008 Sep 30;118(14):1433-41
Fallick C et al. Circ Heart Fail 2011; 4: 669-75.

Using the wrong manufacturer-specific device programmer causes a delay in diagnostic and treatment and can be relevant in these situations.
Techniques to identify a CRMD are following:
- Patient's ID card
- Medical records
- Manufacturers' patient registries (All CRMD manufacturers keep their own in-house registry of patients implanted with their devices and provide 24-hour telephone technical support
- Device specific radiopaque alphanumeric codes (ANC)
All these identification techniques have their problems in clinical practice, and so far no other method or algorithm was available to help out in such a dilemma. Sony Jacob et al. have therefore developed and validated the so-called
Cardiac Rhythm Device Identification Algorithm using X-rays (CaRDIA-X, see below)
The study participants using this algorithm showed an overall accuracy of 96.9%. This study was published in 2011 but only now caught our attention.

Using the chart is a little challenge itself, but very helpful in most cases! Certainly worth keeping in mind!
Jacob S et al. Heart Rhythm. 2011 Jun;8(6):915-22.


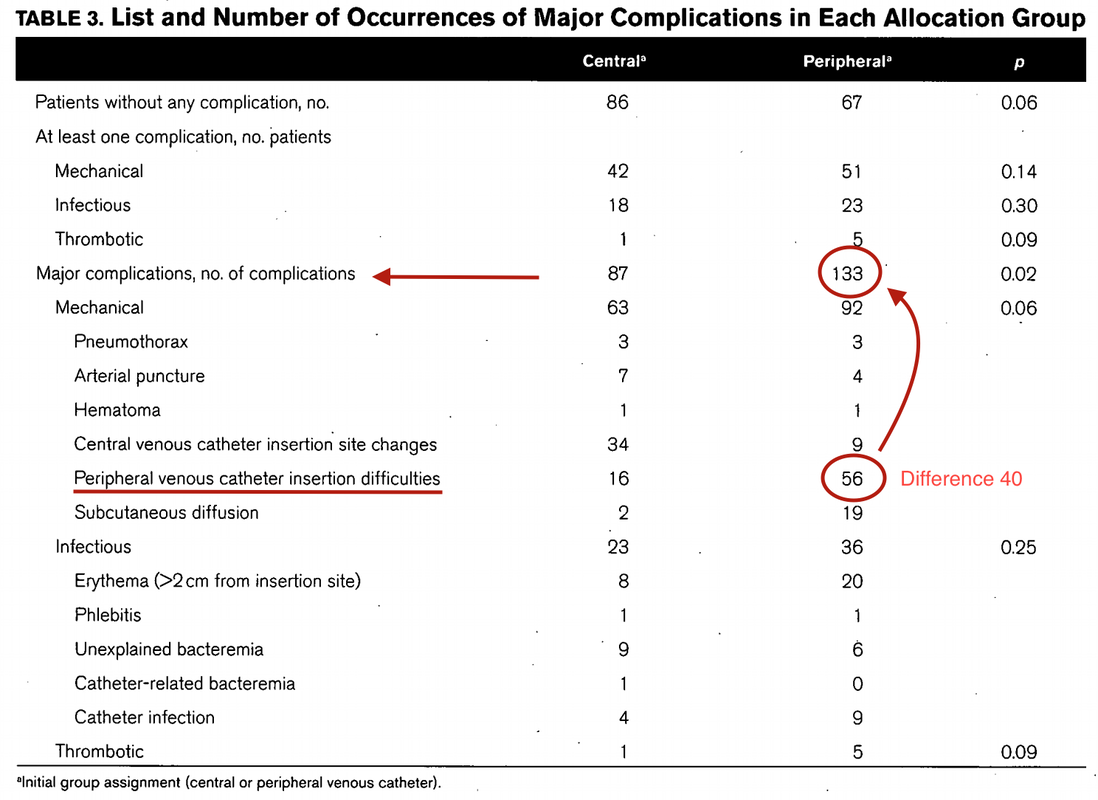
The major problem is that performing a randomized controlled trial to elucidate the true incidence of contrast-induced nephropathy is considered unethical because of the presumption that contrast media administration is a direct cause of acute kidney injury.
While the discussion goes on Hinson et al. have come up with another nice piece of evidence that in emergency situations there is no reason to withhold the application of IV contrast for CT scans when required.
In this single-center retrospective cohort study researchers have included a total of 17'934 patient visits to their emergency department over a period of 5 years. They analysed three patient groups that where demographically similar: contrast-enhanced CT, unenhanced CT and no CT scan performed. Patients were included when their initial serum creatinine level was between 35 umol/L and 352 umol/L. Of all CT scans, 57.2 percent were contrast-enhanced. The probability of developing acute kidney injury was 6.8 percent for patients undergoing contrast-enhanced CT, 8.9 percent for patients receiving unenhanced CT and 8.1 percent for patients not receiving CT at all. This proofs to be the largest controlled study of its kind in the emergency department and shows that:
In current clinical context, contrast media administration for CT scans is NOT associated with an increased incidence of acute kidney injury. And even though a large randomised controlled trial is still missing it seems safe...
To Conclude:
There is no reason to withhold the use of IV contrast media in cases where contrast-enhanced CT is indicated to avoid delayed or missed diagnosis of critical disease.
Hinson J et al. Annals of Emergency Medicine, 2017; DOI: 10.1016/j.annemergmed.2016.11.021 OPEN ACCESS
Crit Cloud Review from 18/01/2015

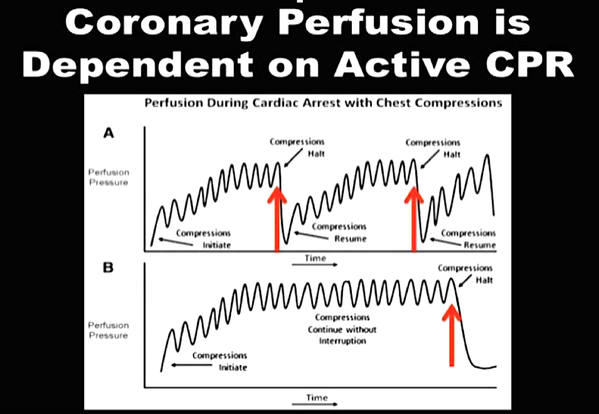
While adrenaline is given for maximum vasoconstriction in order to promote coronary perfusion pressure CPP, amiodarone and sometimes lidocaine are used to promote successful defibrillation of shock-refractory ventricular fibrillation VF or pulseless ventricular tachycardia VT. While the usage of these drugs is undoubtedly very effective in patients with existing circulation the effectiveness during resuscitation remains a matter of debate.
The Effect of Adrenaline
As a matter of fact it has never been proven that adrenalin actually improves long-term outcome. In 2014 Steve Lin and colleagues published a systemativ review on the efficacy of adrenaline in adult out-of-hospital cardiac arrest (OHCA). They were able to show that according to current evidence standard dose adrenaline (1mg) improved rates of survival to hospital admission and return of spontaneous circulation (ROSC) but had no benefit in means of survival to discharge or neurologic outcomes.
What about Amiodarone and Lidocaine?
Kudenchuck et al. now made the effort to look into the efficacy of amiodarone and lidocaine in the setting of OHCA. Used according to the ACLS guidelines 2016 amidarone is given after the third shock applied when treating a shockable rhythm. Two rather small controlled trials have shown so far that using amidarone actually does increase the likelihood of ROSC and the chance to arrive at a hospital alive. It's impact on survival to hospital discharge and neurologic outcome though remains uncertain.
In this randomized, double-blind trial, the investigators compared parenteral amiodarone, lidocaine and saline placebo in adult, non-traumatic, OHCA. They ended up with 3026 patients meeting inclusion criteria and which were randomly assigned to receive amiodarone, lidocaine or saline placebo for treatment. They finally found that neither amiodarone nor lidocaine improved rate of survival to discharge or neurologic outcome significantly. There were also no differences in these outcomes between amiodarone and lidocaine. Across these trial groups also in-hospital care like frequency of coronary catheterisation, therapeutic hypothermia and withdrawal of life-sustaining treatments did not really differ, making a bias due to treatments after admission unlikely.
Take Home
- Amiodarone seems to improve the likelihood of ROSC and survival to hospital admission (similar to adrenaline)
- As there are no other options, I believe amiodarone should remain part of the standard treatment for shockable rhythms in OHCA
- Lidocaine can be safely removed from CPR sets as there is no benefit of over amiodarone
Read here:
N Engl J Med 2016;374:1711-22
Resuscitation, June 2014, Vol 85, Issue 6, p 732-740
New ACLS Guidelines 2015, The Changes
Search
Translate
Select your language above. Beware: Google Translate is often imprecise and might result in incorrect phrases!

Categories
All
Airway
Cardiovascular
Controversies
Endocrinology
Fluids
For A Smile ; )
Guidelines
Infections
Meducation
Neurology
Nutrition
Pharmacology
Procedures
Radiology
Renal
Respiratory
Resuscitation
SARS CoV 2
SARS-CoV-2
Sedation
Sepsis
Transfusion
Archives
January 2021
September 2020
March 2020
February 2020
January 2020
December 2019
November 2019
July 2019
May 2019
March 2019
February 2019
January 2019
December 2018
January 2018
October 2017
August 2017
June 2017
March 2017
February 2017
January 2017
October 2016
July 2016
June 2016
April 2016
February 2016
December 2015
October 2015
September 2015
August 2015
July 2015
June 2015
May 2015
April 2015
March 2015
January 2015
December 2014
November 2014
October 2014
September 2014
August 2014
July 2014
June 2014
May 2014
April 2014
March 2014
February 2014
January 2014
December 2013
November 2013
Author
Timothy Aebi
 RSS Feed
RSS Feed


